
Background
On 21 August 2024 the International Council for Harmonisation (ICH) announced the adoption of the ICH E11A Guideline on Pediatric Extrapolation by Regulatory Members of the ICH Assembly under Step 4. This means the new framework is now ready for implementation by ICH regulators in their respective jurisdictions.
While the existing ICH E11(R1) guideline provides high-level guidance on pediatric extrapolation, sponsors typically had to validate their approaches through regulatory interaction meetings. The ICH E11A guideline builds on the original ICH E11 guideline, which outlines comprehensive principles for the clinical investigation of medicinal products in pediatric populations. With the increasing recognition of the distinct physiological and developmental differences between children and adults, there has been a growing need for more specific and nuanced guidance to address the challenges associated with pediatric clinical trials. This need is particularly critical in rare diseases, as approximately 70% present in childhood, making pediatric extrapolation essential for advancing therapies in these populations. The ICH E11A guideline aims to address these challenges by offering a structured approach to pediatric extrapolation, which refers to the process of applying data from adult or older pediatric populations to younger pediatric subgroups.
Why Was ICH E11A Needed?
Historically, pediatric-specific data has been limited, leading to off-label medication use in children—often resulting in suboptimal or unsafe treatment. One of the key issues identified by the ICH in its 2017 concept paper was the 7- to 10-year gap between the initial adult approval and the inclusion of pediatric-specific information in product labeling.
By providing clearer methodologies and standardized criteria for pediatric extrapolation, ICH E11A aims to:
- Improve the ethical and scientific integrity of pediatric drug development
- Ensure children have access to safe and effective therapies based on robust extrapolation data
- Reduce reliance on off-label prescribing by leveraging extrapolated data to support approved pediatric dosing and treatment labeling
This blog explores key features of ICH E11A, the role of modelling and simulation, and the implications of this guidance for sponsors conducting studies in the US and the EU.
Existing Guidance on Pediatric Extrapolation
With the release of ICH E11A, there is now greater global alignment on pediatric extrapolation approaches, which should streamline development and expedite drug approvals for pediatric populations.
Before the introduction of ICH E11A, multiple guidelines existed to inform pediatric extrapolation in clinical trials. Key guidance in place from regulatory authorities included:
- EMA’s adopted Reflection Paper on the Use of Extrapolation in the Development of Medicines for Paediatrics (2018)
- EMA’s Structured Guidance on the Use of Extrapolation (2022)
- ICH E11(R1) Guideline on Clinical Investigation of Medicinal Products in Pediatric Population (2017)
With the release of ICH E11A, there is now greater global and regulatory alignment on pediatric extrapolation approaches across ICH regions, which should streamline development for sponsors and expediate drug approvals for pediatric populations.
Key Features of ICH E11A
ICH E11A largely retains the content from its Step 2 draft (April 2022), reinforcing its core objective: accelerating the availability of pediatric medicines. To achieve this, the new guideline outlines a stepwise approach for sponsors to:
- Understand the key differences between the pediatric and adult populations
- Use this knowledge to develop a structured extrapolation plan
- Execute the plan using appropriate modeling, simulations, and study designs
The foundation of this approach lies in the first step—understanding key differences between pediatric and adult populations. This evaluation forms what is known as the pediatric extrapolation concept, which serves as the basis of an extrapolation plan.
The following sections explore each of these steps in detail, beginning with the development of a pediatric extrapolation concept.
1. Development of a Pediatric Extrapolation Concept
Before developing a pediatric extrapolation plan or designing any necessary studies, sponsors must first evaluate how the adult and pediatric populations differ across three key areas: disease characteristics, drug pharmacology, and treatment response. This evaluation—referred to as the ‘pediatric extrapolation concept’—helps determine what is known and unknown in each of these areas and serves as the foundation for the extrapolation plan.
Differences in Disease: The guidance shifts from a binary ‘yes’ or ‘no’ definition to a continuum, recognizing varying degrees of difference. This analysis should consider several key factors, including:
- Pathophysiology of the disease
- Disease definition and diagnostic criteria
- Disease progression over time
Differences in Drug Pharmacology: Key clinical pharmacology considerations must account for absorption, distribution, metabolism, and excretion (ADME) differences in pediatrics, particularly during maturation, as well as any variations in the drug’s mechanism of action (MOA).
Differences in Treatment Response: This may include assessing data on exposure-response relationships in adults versus pediatrics, as well as evaluating drug target similarities and differences between both age groups.
The new guideline also provides long-awaited clarity on safety extrapolation in pediatrics, which is generally more challenging than efficacy extrapolation. The guideline suggests that safety extrapolation “could be considered” in certain circumstances, provided that robust clinical trial and animal study data support its feasibility. If there are substantial differences in the expected safety profile—such as potential on- or off-target effects in pediatrics or risks to pediatric development —extrapolation may not be possible, and additional safety data may be required to address these gaps.
The guideline also outlines the various data types that may be used to support the pediatric extrapolation concept. Notably, real-world data (RWD) is now recognized as a potential tool. Sponsors are advised to engage with regulators to discuss how RWD could be used to support pediatric extrapolation.
2. Development of a Pediatric Extrapolation Plan
Once sponsors establish a complete understanding of the similarities and differences between the target (pediatrics) and reference (adult) populations, established by the extrapolation concept, they can develop a structured extrapolation plan. This plan may include:
- Additional clinical studies that must be conducted to address data gaps between the reference and target populations
- Relevant data analyses, including model-based analyses, to support extrapolated findings
A notable recommendation in Section 4.1.1 of the guideline is the strong suggestion to:
- Include adolescent subjects in adult clinical trials, or
- Study adolescents in parallel when disease characteristics, drug pharmacology, and treatment response are sufficiently similar between adolescent and adult participants
This recommendation builds on the guidelines in ICH E11(R1) which advises that for serious diseases affecting both adults and pediatrics, pediatric medicinal product development should begin early, following initial adult assessments. The strengthening of this position contrasts with common past practices where adult development was often completed first, delaying pediatric studies.
A key theme in ICH E11A is the use of Model-Informed Drug Development (MIDD). Below, we outline critical modeling approaches used in pediatric drug development, along with a case example illustrating its application.
3. Role of Modeling and Simulation in Pediatric Drug Development
Modeling and Simulation Approaches – Modeling and simulation plays a vital role in the development of pediatric trials. It provides the opportunity to:
- Help address ethical constraints in pediatric trials and provide data-driven analyses to overcome these limitations
- Ensure age-appropriate dosing, factoring in pediatric maturation
- Determine optimal sample size and time points for PK/PD characterization
- Reduce risks by testing hypotheses before conducting pediatric trials
Most importantly, modeling and simulation enables data-driven dose selection, identifying doses most likely to succeed in a pediatric clinical setting while highlighting areas of uncertainty. Extrapolation from PK/PD modeling in adults and/or adolescents helps predict pediatric clinical trial outcomes. Additionally, as pediatric trials progress or conclude, initial modelling assumptions can be validated or refined based on real pediatric data.
Dose selection – Selecting the right dose before a trial begins, is critical to ensure both safety and efficacy. This process involves:
- Accounting for ADME differences, including the impact of pediatric maturation in metabolism and/or excretion, while also considering known efficacy markers
- Addressing challenges in characterizing pediatric efficacy, using exposure matching from a reference population (adults or adolescents) when direct efficacy in pediatrics is not available
- Leveraging simulations, including hypothetical populations, to determine optimal dose levels and dosing schedules, assess target attainment, evaluate potential safety events driven by exposure, and integrate PK/PD modeling to refine dosing strategies
- Assessing exposure-based safety risks and adjusting dosing paradigms to minimize adverse events while optimizing efficacy
Biomarkers – Biomarkers can be used as surrogate endpoints in pediatric extrapolation. Key considerations include:
- Applicability – The chosen biomarker should be relevant to both the reference (adult) and target (pediatric) populations
- Time course – Establishing how the biomarker time course changes over disease progression and treatment duration is essential
- Quantifiable relationship – If a clear correlation exists between the biomarker and clinical efficacy, models can be used to support extrapolation
- Predictive modeling – Simulations can predict biomarker responses under different dosing paradigms before testing in pediatric patients
By integrating validated biomarkers into modeling approaches, sponsors can improve pediatric dosing predictions while minimizing the need for extensive clinical trials.
Exposure matching – Exposure matching is the most common application of modeling and simulation for pediatric extrapolation. Key steps include:
- Establishing reference exposure ranges – Using exposures from the reference population that have been modeled to correlate with safety concerns and efficacy targets
- Adjusting for pediatric-specific factors – Models are updated to account for pediatric differences, including allometric scaling for body size and maturation functions affecting drug metabolism
- Simulating expected exposures – These adjusted models predict drug exposures in pediatric populations, refining dose selection
- Accounting for interindividual variability – Models incorporate variability to ensure dosing recommendations reflect real-world differences
- Selecting hypothetical populations – Ensuring simulated populations accurately reflecting both the target pediatric demographic and what is feasible for a pediatric clinical trial.
- Running trial simulations – Testing dose levels and schedules through trial simulations to determine the most appropriate pediatric dose for clinical evaluation
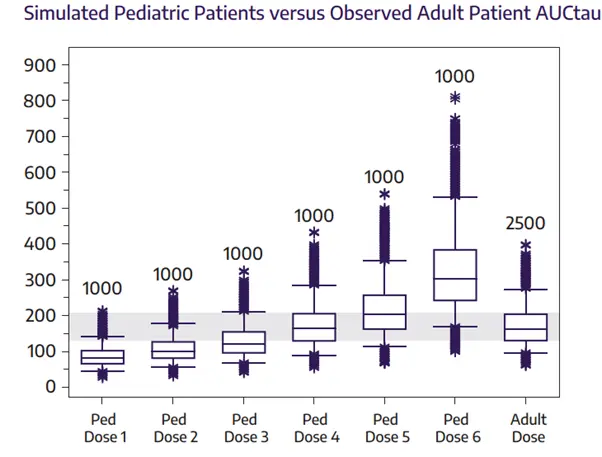
Final exposures are compared to the reference population to select the most appropriate dose to evaluate in a clinical trial setting. The example below illustrates this process: a simulation was performed where a specific pediatric age group was exposed to various dose levels through simulation, with each dose level assessed for its alignment with adult reference exposures. In this scenario, Ped Dose 4 was chosen as it most closely matched the reference adult exposure, ensuring an appropriate pediatric dose selection.
What does this mean for sponsors involved in pediatric development?
The adoption of ICH E11A means global regulatory alignment on expectations for pediatric development and the use of pediatric extrapolation.
Key Consideration for Sponsors
With a strong push to include adolescents in adult development programs, sponsors may need to conduct these trials. When doing so, key considerations include:
- Pediatric-specific study sites to ensure appropriate recruitment and trial execution.
- Parental consent and adolescent assent to ensure ethical and regulatory compliance.
- Assessing the suitability of safety and efficacy endpoints to determine whether the same measures can be across adolescent and adult populations.
Beyond adolescent inclusion, broader considerations for pediatric development include:
- Leveraging model-informed drug development approaches to support extrapolation and optimize study design, as encouraged by the new guideline.
- Harmonization of regulatory requirements under the ICH guideline, potentially reducing region-specific differences and promoting a more unified approach to pediatric extrapolation.
Final Thoughts: Moving Forward with ICH E11A
ICH E11A marks a significant step forward in pediatric drug development, offering clearer methodologies and stronger regulatory alignment across ICH regions. By integrating model-based approaches, sponsors can improve trial design, dose selection, and extrapolation accuracy—an approach particularly important in rare diseases, where pediatric patients make up the majority of cases, and clinical trial data is often limited. The shift toward earlier inclusion of adolescents in adult studies presents both challenges and opportunities. To stay ahead of evolving regulatory expectations, sponsors should begin incorporating ICH E11A principles now, engaging with regulators early and leveraging model-informed drug development to optimize their pediatric programs. Ultimately, ICH E11A is expected to enhance the availability of safe, effective pediatric therapies worldwide.
If you’re interested in learning more about pediatric drug development, read our related blog, Leveraging Modeling and Simulation in Pediatric Drug Development. To learn how our experts can support your regulatory strategy and modeling efforts, visit Regulatory Consulting Services and Modeling & Simulation Services.
About the Authors
John McIntyre PhD is Director, Clinical Strategy at Allucent, specializing in regulatory strategy, clinical development, and medical writing. With over 15 years of experience, he has deep expertise in EU regulatory procedures, orphan drug designations, pediatric plans, and pharmacovigilance. A former deputy-QPPV, he has guided sponsors through clinical strategy and marketing authorization submissions. He holds a PhD in Biochemistry and Genetics from University College London and a Bachelor’s in Biotechnology from NUI Galway.
Jessica K. Roberts PhD, MSCI is Senior Director, Pharmacometrics at Allucent, specializing in clinical pharmacology and pharmacometrics. She holds a PhD in Pharmacology and Toxicology and a Master’s in Clinical Investigation from the University of Utah. Her experience includes neonatal/pediatric anti-infectives at Primary Children’s Hospital, oncology clinical trials and gene therapy development at St. Jude Children’s Research Hospital and consulting roles at Metrum Research Group and Cognigen/Simulations Plus.
Share This